ABC de Fertilidad
Causas de la Infertilidad
Inicia Tu Camino
Quiero tener un Bebé
Quiero Congelar / Preservar
Quiero Donar
Estoy Embarazada
Tratamientos
- Primera Consulta
- Fertilización In Vitro
- FIV con ICSI
- FIV con PICSI
- FIV ABC (Mini FIV)
- FIV Método ROPA
- Análisis Genético
- Soluciones FIV Multiciclo (BEC)
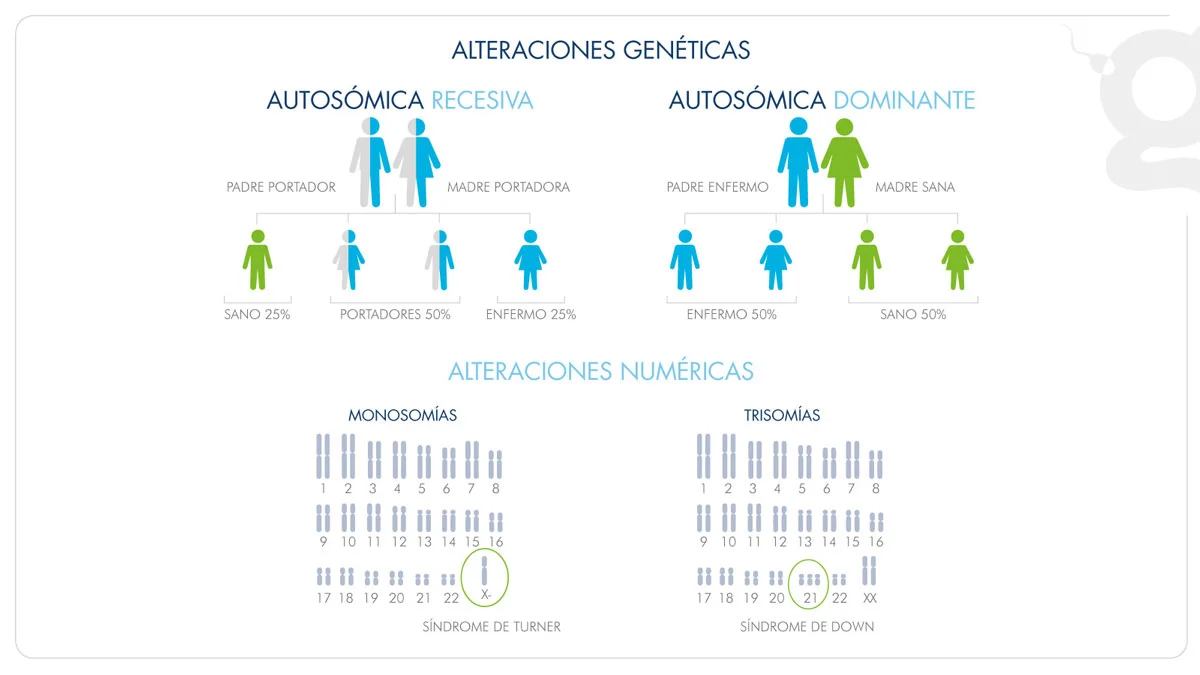
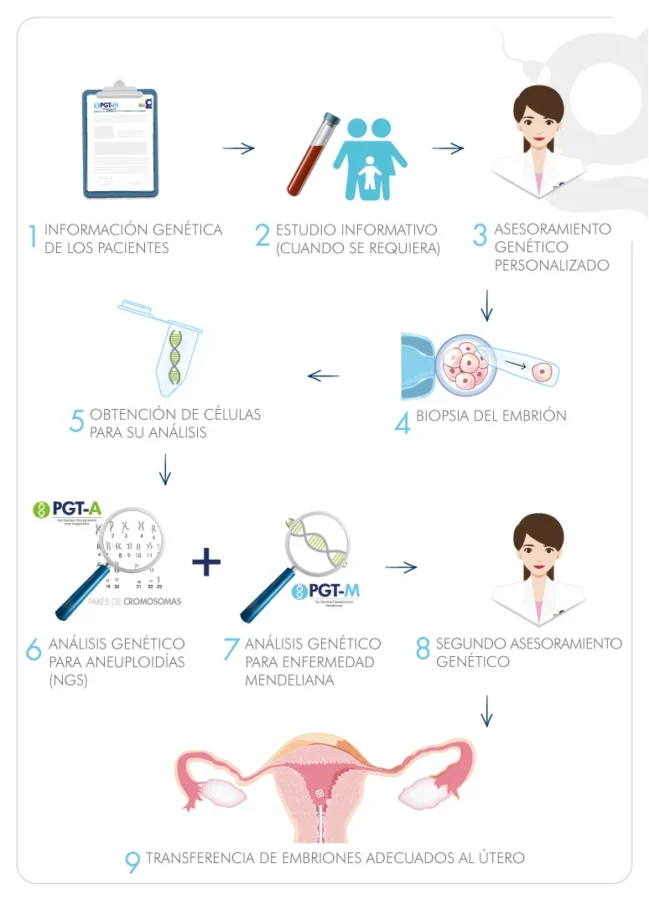
- PGT-A
- PGT-M
- Selección de Sexo
- Regeneración Ovárica
- Regeneración Endometrial
- Preservación de Óvulos
- IVF MORE®
- Subrogación
- Score de implantación
- Test Prenatal no Invasivo
- Banco de Semen Internacional
- Unidad Materno Fetal
- Salud Emocional
- Inseminación Artificial
- Donación de Óvulos
- Donación de Esperma